By: Casey Doyle
Clinical Technologies PA Group Leader
What impact can IRT have on my supply chain structure?
There are many challenges and pressures within clinical trials. Some, if not most, of those challenges and pressures can be relieved through optimizing clinical supply within a validated IRT solution. From depot management to accountability and reconciliation efforts, IRT systems are designed with the flexibility to automate steps, provide continuity, and eliminate concerns for patient dosing and site stock outs.
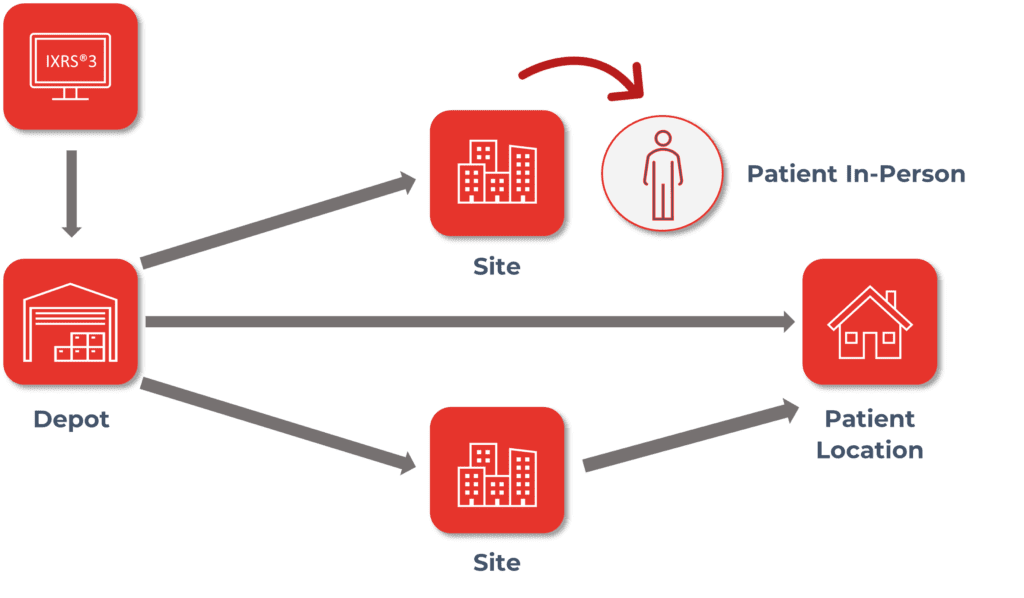
Supply chain can be a delicate dance between managing the needs of sites, depots, vendors, etc. The more sites, depots, and kit types to manufacture and distribute, the more cumbersome it becomes to effectively manage needs across multiple countries in a variety of time zones. IRT systems can help you seamlessly integrate your different supply chain models to meet the needs of your clinical trial by providing the flexibility of multiple resupply strategies, automated drug ordering, shipping for temperature sensitivities, alerting for expiry and low stock, inventory at a glance through blinded and unblinded reporting, tracking, and return of reconciled supplies and so much more.
An IRT system can additionally incorporate capabilities such as drug pooling which can provide plasticity of inventory across a study by consolidating supply and streamlining management of depot activities. This can be particularly useful for program studies that have very high-cost materials or for those forecasting potential manufacturing delays due to resourcing of material. Drug Pooling allows supply to be shared across multiple protocols with lesser need for multiple manufacturing runs as well as reducing the carbon footprint of your trial(s) through the reduction of depot transfers.
What efficiencies in logistics can an IRT system provide?
One of the more difficult pieces of clinical trials can be ensuring that your clinical supply shows up where it is needed and when it is needed with a minimal number of disruptions. An IRT solution can help take care of both the “where” and “when” additionally providing data visibility to “how” clinical supply is being used.
IRT systems can automatically determine where clinical supplies are needed and raise shipments for clinical sites based on various factors such as static values, buffers, titrations, projected visits, damaged supply, etc. While the majority of when a shipment arrives at the site is impacted by the distributor, an IRT system can indicate to sites and clients an estimated delivery date based on calculated shipping estimates as well as provide delivery tracking capabilities through customized data integrations. This helps sites more accurately manage their subject’s visit schedule and ensure that supply will be available when the subjects are on site. Clients and sites have the ability to monitor how supply is being used through real-time reporting within an IRT. Reports allow clinical teams to monitor subject visits including completions and discontinuations, as well as, to monitor supply at sites as well as across depots.
What communication capabilities does an IRT offer for supply chain management?
With an IRT solution, there is no longer the need to play middleman with 3rd party depots. Drug order forms can be customized to meet the needs of your distributors and are automatically sent to specified depot contacts upon order creation. With advancements in technology and expanding needs for faster delivery, depot integrations are another way in which shipment communications and inventory updates can be managed shortening the time it takes to facilitate orders from depot to site.
IRT also provides alerting which can be customized to the needs of the trial. Alerts can be transactionally triggered based on events, for example, acknowledgment of a shipment that was outside of temperature range. Additionally, alerts can be configured to trigger at various thresholds such as when depot inventory falls below a certain count, or if a depot has completely stocked out, etc. Alerts can be formed to meet a variety of scenarios and provide a different way of monitoring milestones without continuously being connected to the IRT application.
What if I have unique or highly complex trial needs?
The leaders in IRT understand the complexities of modern clinical trials and can customize your IRT to meet any unique needs. From multi-depot ordering to drug pooling to pre-enrollment drug reservation and beyond, there is an IRT solution to fit your trial’s demands.
Is it worth my budget to consider using an IRT?
Absolutely! An IRT solution is an investment in your clinical trial. Not only is it a time save at the end of the trial for reporting data pulled directly from the IRT as well as from not having to manage a paper trail, but it additionally saves time for clinical staff and patients who just want the most efficient way of participating. An IRT system allows sponsors and clinical sites to focus on what is most important, the patient. The use of an IRT system allows you to more effectively plan and adapt. Protocol changes happen and they happen frequently in some cases. IRT systems can be flexible and provide the tools to help you adapt your supply to meet the needs of the trial in addition to allowing study teams to make informed decisions in a timely, controlled manner. Knowing the industry and the ever-changing variables, there can’t be a one-size-fits-all solution. Leading IRT vendors including Almac Clinical Technologies offer multiple levels of IRT solutions to fit a variety of budget needs.